

Ihr ausführlicher Leitfaden zu den Bestimmungen für Medizinprodukte im Vereinigten Königreich
Medizinprodukte sind sehr vielfältig. Sie umfassen ein breites Spektrum von Produkten, von chirurgischen Masken über Beatmungsgeräte bis hin zu Herzkathetern. So unterschiedlich diese Produkte auch sind – sie haben eines gemeinsam: Zum Schutz des Patienten unterliegen sie je nach ihrer Klassifizierung strengsten Qualitätsstandards und spezifischen gesetzlichen Anforderungen.
Für Hersteller, die ihr Produkt auf den Markt bringen wollen, ist es daher unerlässlich, den rechtlichen Rahmen eines bestimmten Absatzmarktes zu verstehen. Auf dieser Website konzentrieren wir uns auf das regulatorische Umfeld für Medizinprodukte im Vereinigten Königreich. Seit dem Austritt aus der Europäischen Union gab es im Vereinigten Königreich erhebliche Veränderungen, insbesondere durch die Einführung der UKCA-Kennzeichnung (United Kingdom Conformity Assessed) und der UKNI-Kennzeichnung (United Kingdom Northern Ireland).
UKCA – Vier Buchstaben, die neue Türen öffnen
Die britische Regierung hat das UKCA-Zeichen als eindeutigen Nachweis dafür eingeführt, dass ein Produkt den geltenden Rechtsvorschriften entspricht. Es deckt die meisten Produkte ab, für die zuvor die CE-Kennzeichnung erforderlich war. Deshalb entsprechen die gesetzlichen Anforderungen für die UKCA-Kennzeichnung bei vielen Produktgruppen nach wie vor den Anforderungen der CE-Zertifizierung. Es gibt jedoch vermehrt spezifische Vorschriften im Vereinigten Königreich, die von den EU-Vorschriften abweichen und von Herstellern und Händlern berücksichtigt werden müssen.
Für Hersteller von Medizinprodukten, In-vitro-Diagnostika und aktiven implantierbaren medizinischen Geräten ist diese Kennzeichnung von entscheidender Bedeutung, da sie belegt, dass ihr Produkt den geltenden UK-Vorschriften entspricht. Für diese Produktgruppe ist eine UKCA-Kennzeichnung vorgeschrieben und erfordert mitunter sogar die Einschaltung einer im Vereinigten Königreich zugelassenen Stelle (UK Approved Body).
Die Anforderungen sind in der Medizinprodukteverordnung 2002 (SI 2002 Nr. 618, in der jeweils geltenden Fassung) (UK MDR) definiert. Die zugrundeliegenden europäischen Richtlinien, auf die in der UK MDR Bezug genommen wird, decken unterschiedliche Aspekte der Anforderungen an das Medizinprodukt ab, einschließlich seiner Auslegung, Herstellung und Kennzeichnung sowie der Informationen, die zusammen mit dem Produkt geliefert werden müssen. Im folgenden Abschnitt werden wir uns die UK MDR genauer ansehen.
Es ist zu beachten, dass die britische Regierung den Übergangszeitraum (Übergangsregelungen) von der CE- zur UKCA-Kennzeichnung für viele Produktgruppen verlängert hat, um Engpässe bei Medizinprodukten auf dem britischen Markt zu vermeiden. Im Abschnitt „Übergangsregelungen“ finden Sie Einzelheiten zu verschiedenen Arten von Medizinprodukten. Allerdings müssen alle Medizinprodukte, die in Großbritannien (England, Schottland, Wales) in Verkehr gebracht werden, seit 2021 bei der Medicines and Healthcare products Regulatory Agency (MHRA) registriert werden. Auch bestimmte Medizinprodukte, die in Nordirland in Verkehr gebracht werden (einschließlich IVDs, maßgefertigte Produkte und Systeme sowie Behandlungseinheiten), müssen bei der MHRA registriert werden.
Das UKCA-Zertifizierungssystem – Wer sind die Beteiligten?
MHRA
Die Medicines and Healthcare products Regulatory Agency ist die staatliche Behörde, die dafür zuständig ist, dass Medizinprodukte und andere medizinische Erzeugnisse ordnungsgemäß funktionieren und eine angemessene Sicherheit aufweisen. Die MHRA spielt eine wichtige Rolle bei der Regulierung von Medizinprodukten im Vereinigten Königreich und beaufsichtigt den gesamten Lebenszyklus vom Produktdesign über die Herstellung und klinische Bewertung bis hin zur Überwachung nach dem Inverkehrbringen.
Die Behörde ist außerdem für die Benennung und Überwachung von Organisationen zuständig, die die Zertifizierung gemäß der UK MDR durchführen. Dies sind die so genannten UK Approved Bodies.
Hinweis: Akkreditierungen für andere Produktgruppen wie PSA, Maschinen, Spielzeug usw. werden von UKAS, dem United Kingdom Accreditation Service, ausgestellt.
UK Approved Body
Ein UK Approved Body ist eine Organisation, die von der MHRA zur Durchführung einer bestimmten Konformitätsbewertung benannt wurde, um die Einhaltung der Rechtsvorschriften zu gewährleisten. Die Benennung einer Organisation als UK Approved Body umfasst in der Regel ein formelles Bewerbungs- und Evaluierungsverfahren, bei dem die Organisation ihre Kompetenz, ihr Fachwissen, ihre Ressourcen und ihre Fähigkeit zur Erfüllung der benannten Funktionen oder Aufgaben unter Beweis stellen muss.
Wir freuen uns, Ihnen mitteilen zu können, dass TUV Rheinland UK Ltd. von der MHRA als UK Approved Body gemäß UK MDR benannt wurde.
Hinweis: Frühere Benannte Stellen mit Sitz im Vereinigten Königreich sind UK Approved Bodies geworden. Sie können nicht länger CE-Zertifikate für den EU-Markt ausstellen, da sie von der EU nicht anerkannt werden.
UK Responsible Person
Die UK MDR führt die Rolle einer UK Responsible Person ein, eines verantwortlichen Vertreters, der im Namen eines außerhalb des Vereinigten Königreichs ansässigen Herstellers agiert.
Auf diese Weise soll gewährleistet werden, dass es eine juristische Person mit Sitz im Vereinigten Königreich gibt, die für die Konformität des Produkts verantwortlich gemacht werden kann. Daher müssen außerhalb des Vereinigten Königreichs ansässige Hersteller eine UK Responsible Person per schriftlichem Mandat ernennen, in dem die spezifischen Aufgaben, für die diese Person verantwortlich ist, niedergeschrieben werden müssen. Dies ist notwendig, um ihr Produkt im Vereinigten Königreich auf den Markt zu bringen.
Die wichtigsten Zuständigkeiten sind:
- Gewährleistung der Durchführung des Konformitätsbewertungsverfahrens
- Gewährleistung der Verfügbarkeit der technischen Dokumentation und der Konformitätserklärung auf Anfrage seitens der MHRA
- Beantwortung aller Anfragen der MHRA
- Führung eines Verzeichnisses über Beschwerden, nicht-konforme Produkte und Produktrückrufe sowie Bereitstellung der entsprechenden Informationen
- Meldung schwerwiegender Vorfälle an die MHRA
Letztlich bleibt jedoch der Hersteller für die Konformität seines Produkts verantwortlich, auch wenn er Aufgaben an eine UK Responsible Person delegiert hat.
Hersteller
Der Hersteller (oder Importeur) eines Medizinprodukts ist verpflichtet, dafür zu sorgen, dass das Produkt in Übereinstimmung mit den geltenden Anforderungen der einschlägigen Rechtsvorschriften entwickelt und hergestellt wurde. Das bedeutet, dass der Hersteller auch für die Durchführung einer Konformitätsbewertung verantwortlich ist, unabhängig davon, ob eine Selbsterklärung ausreicht oder eine obligatorische externe Konformitätsbewertung durch eine unabhängige Stelle erforderlich ist. Der Hersteller muss eine technische Dokumentation anfertigen und eine Konformitätserklärung für das Vereinigte Königreich ausstellen und beide Unterlagen 10 Jahre lang aufbewahren. Er ist außerdem verpflichtet, das Produkt ordnungsgemäß zu kennzeichnen.
Hinweis: Dies gilt auch für Importeure oder Vertriebshändler, die ein Produkt unter ihrem Namen und ihrer Marke vertreiben, da sie in der Regel als Hersteller gelten.
Weitere Informationen über die Zuständigkeiten der verschiedenen Parteien finden Sie auf der folgenden Website der britischen Regierung.

Wo gilt die UKCA-Kennzeichnung?
Auf den ersten Blick erscheint es offensichtlich, dass die UKCA-Kennzeichnung für Produkte gilt, die im Vereinigten Königreich vertrieben werden. Wir möchten jedoch auf einige Sachverhalte hinweisen, die häufig zu Verwirrung führen.
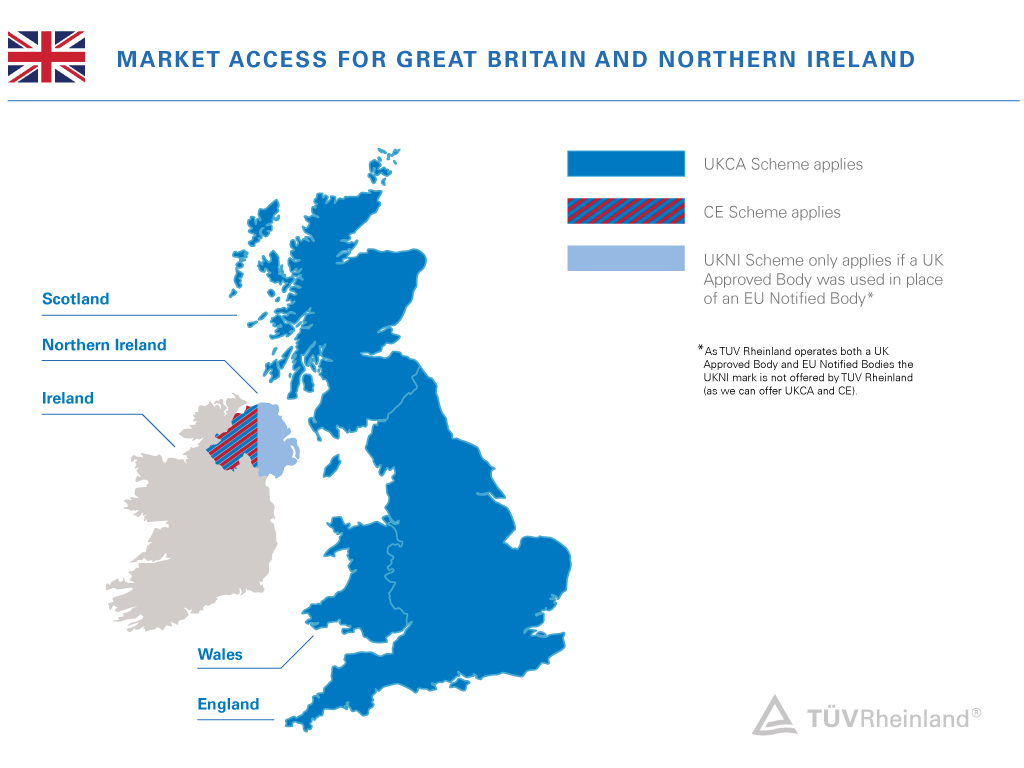
Wenn vom Vereinigten Königreich die Rede ist, ist damit der Zusammenschluss von vier Ländern gemeint: England, Wales, Schottland und Nordirland. Großbritannien hingegen umfasst nur England, Wales und Schottland.
Für Nordirland mit seiner Binnengrenze zu Irland (und damit zur Europäischen Union) gelten besondere Regelungen, die im Nordirland-Protokoll festgehalten sind.
Seit 2022 gelten in Nordirland beide EU-Verordnungen: die EU MDR (Verordnung 2017/745) und die EU IVDR (Verordnung 2017/746).
Das bedeutet, dass die UK MDR und das UKCA-Zertifizierungssystem nur für die Länder Großbritanniens gelten und der Markt in Nordirland eine CE-Kennzeichnung erfordert.
CE- und UKNI-Kennzeichnung für Nordirland
Für das Inverkehrbringen eines Produkts in Nordirland ist die CE-Kennzeichnung erforderlich, die entweder nach einer Selbsterklärung vom Hersteller angebracht oder von einer EU-Benannten Stelle durch eine Behörde eines EU-Mitgliedstaats gemäß der EU MDR oder der EU IVDR vergeben werden kann. Wenn ein Medizinproduktehersteller eine von der EU anerkannte Benannte Stelle für die obligatorische externe Konformitätsbewertung einsetzt, reicht die CE-Kennzeichnung allein aus, um ein Produkt in Nordirland in Verkehr zu bringen.
Wenn jedoch eine im Vereinigten Königreich anerkannte Stelle (UK Approved Body) die obligatorische externe Konformitätsbewertung durchführt, ist die UKNI-Kennzeichnung erforderlich. In diesen Fällen dürfen die Hersteller von Medizinprodukten die UKNI-Kennzeichnung nicht einzeln anbringen. Diese muss immer mit einer CE-Kennzeichnung einhergehen. Produkte mit der kombinierten „CE & UKNI“-Kennzeichnung werden auf dem EU-Markt nicht akzeptiert.
Es ist zu beachten, dass sich die Regeln und Ausnahmen für Nordirland im Laufe der Verhandlungen zwischen dem Vereinigten Königreich und der Europäischen Union erheblich verändert haben.
Hier sind einige aktuelle Unterschiede zwischen Großbritannien und Nordirland:
- Doppelte Kennzeichnung: In Nordirland werden sowohl die UKNI-Kennzeichnung als auch die CE-Kennzeichnung akzeptiert. Während die CE-Kennzeichnung allein ausreicht, um ein Produkt auf den Markt zu bringen, muss die UKNI-Kennzeichnung immer von einem CE-Zeichen begleitet werden.
- UKNI-Kennzeichnung: Einige Produkte, für die spezifische EU-Rechtsvorschriften gelten und die als „relevante nordirische Waren“ ausgewiesen sind, müssen die UKNI-Kennzeichnung tragen. Diese Kennzeichnung wird für Produkte verwendet, die in Nordirland hergestellt oder nach Nordirland eingeführt werden und für den Verkauf in Nordirland oder sowohl in Nordirland als auch auf dem EU-Markt bestimmt sind.
- Konformitätsbewertungsstellen: Hersteller in Nordirland können weiterhin die von der EU anerkannten Konformitätsbewertungsstellen (Conformity Assessment Bodies, CAB) nutzen. Dies ist von Bedeutung für Hersteller, die eine externe Konformitätsbewertung für Medizinprodukte mit höheren Risiken benötigen.
- Konformitätserklärung für Nordirland: Hersteller, die Produkte auf dem Markt in Nordirland in Verkehr bringen, sollten eine Konformitätserklärung für Nordirland ausstellen. Aus dieser Erklärung sollte hervorgehen, dass das Produkt den geltenden Vorschriften des Vereinigten Königreichs bzw. der EU entspricht, je nachdem, für welchen Markt es bestimmt ist.
- MHRA-Registrierung: Alle im Vereinigten Königreich (Großbritannien oder Nordirland) vertriebenen Medizinprodukte benötigen eine MHRA-Registrierung. Alle maßgefertigten Medizinprodukte müssen innerhalb von 28 Tagen nach ihrer Einführung auf dem Markt in Nordirland bei der MHRA registriert werden.
Die UK MDR 2002
Wie bereits erwähnt, ist die Medizinprodukteverordnung (UK MDR) 2002 (SI 2002 Nr. 618, in der jeweils geltenden Fassung) ein wichtiges Regelwerk, das die Herstellung und den Vertrieb von Medizinprodukten im Vereinigten Königreich regelt. Für Hersteller ist es unerlässlich, diese Verordnung zu verstehen, da sie die Anforderungen festlegt, die erfüllt werden müssen, um ein Medizinprodukt im Vereinigten Königreich legal in Verkehr zu bringen.
Mit der UK MDR wurden drei Richtlinien im Vereinigten Königreich gesetzlich verankert:
- EU-Medizinprodukte-Richtlinie 93/42/EWG (MDD)
- Richtlinie über aktive implantierbare medizinische Geräte 90/385/EWG (AIMDD)
- Richtlinie über In-Vitro-Diagnostika 98/79/EG (IVDD)
Wenn von Medizinprodukten im Zusammenhang mit der UKCA-Zertifizierung die Rede ist, schließt dies daher auch immer In-vitro-Diagnostika und aktive implantierbare medizinische Geräte ein.
Da die UK MDR auf diesen europäischen Richtlinien basiert, beruhen die Anforderungen an die UKCA-Kennzeichnung auf den aus der EU-Gesetzgebung abgeleiteten Anforderungen. Mit dem Austritt des Vereinigten Königreichs aus der Europäischen Union wurde die UK MDR jedoch so geändert, dass sie nun als eigenständige Richtlinie gilt.
Die UK MDR deckt ein umfangreiches Themenspektrum ab, darunter die Klassifizierung von Medizinprodukten, die Konformitätsbewertungsverfahren, die Rolle der UK Approved Bodies, die Überwachung nach dem Inverkehrbringen und die Anforderungen an die klinische Bewertung. Außerdem werden verschiedene Schlüsselbegriffe und Abkürzungen eingeführt, die in der Verordnung verwendet werden.
Einer der wichtigsten Aspekte der UK MDR sind die Konformitätsbewertungsverfahren. Hersteller müssen diese Verfahren befolgen, um nachzuweisen, dass ihr Medizinprodukt den Anforderungen der Verordnung entspricht. Je nach Medizinprodukt und dessen Klassifizierung muss ein bestimmtes Verfahren gewählt werden. Medizinprodukte, die als risikoreicher klassifiziert werden, erfordern in der Regel eine strengere Bewertung.
In der UK MDR werden darüber hinaus die Anforderungen an die Durchführung von Audits, die Qualifizierung von Personal und die Kontrolle von Dokumenten und Aufzeichnungen festgelegt. Mit diesen Anforderungen soll sichergestellt werden, dass die Hersteller über zuverlässige Qualitätsmanagementsysteme verfügen und ein hohes Maß an Kontrolle über ihre Prozesse und Produkte aufrechterhalten.
Die wichtigsten Unterschiede zwischen der EU MDR und der UK MDR
Obwohl die EU MDR und die UK MDR auf den gleichen Richtlinien beruhen, gibt es einige wichtige Unterschiede zwischen diesen beiden Verordnungen. Hersteller, die beide Märkte beliefern, müssen die Vorgaben beider Verordnungen sehr genau beachten und separate Zertifizierungen gemäß der jeweiligen Verordnung beantragen.
Hier sind die Hauptunterschiede:
- Geltungsbereich: Grundsätzlich gelten die EU MDR und die EU IVDR in allen Mitgliedsstaaten der Europäischen Union und in Nordirland, während die UK MDR nur in Großbritannien gilt. Es ist zu beachten, dass die Übergangsfrist noch andauert. Die CE-Kennzeichnung wird im Vereinigten Königreich weiterhin anerkannt und akzeptiert. Die Ablauffrist hängt von der Klassifizierung des Medizinprodukts ab.
- Zertifizierung: Für die Zertifizierung von Medizinprodukten gelten unterschiedliche Konformitätsbewertungsverfahren. Die EU MDR/EU IVDR nutzt die EU-Benannte Stelle (EU Notified Body), wohingegen die UK MDR die im Vereinigten Königreich anerkannte Stelle (UK Approved Body) für die entsprechenden Konformitätsbewertungsverfahren nutzt.
- Produktgruppen: Die UK MDR gilt für allgemeine Medizinprodukte, aktive implantierbare medizinische Geräte und In-vitro-Diagnostika (IVDs). Für den EU-Markt sind die Anforderungen separat in der EU MDR und der EU IVDR definiert.
- UKCA-Kennzeichnung: Für den britischen Markt ist eine andere Kennzeichnung auf dem Produkt und/oder der Verpackung erforderlich. Die Kennnummer der im Vereinigten Königreich anerkannten Stelle (UK Approved Body) muss auf dem Produktetikett aufgeführt sein. Um den Herstellern die Umstellung zu erleichtern, müssen Produkte mit einer CE-Kennzeichnung nicht umetikettiert werden, und auch die doppelte Kennzeichnung wird bis zum Ende der Übergangszeit weiterhin akzeptiert (Übergangsregelungen).
- UKCA Responsible Person: Wie bereits weiter oben erläutert, müssen Hersteller, die nicht im Vereinigten Königreich ansässig sind, eine verantwortliche Person mit Sitz im Vereinigten Königreich benennen, damit gewährleistet ist, dass es eine juristische Person im Vereinigten Königreich gibt, die für die Konformität des Produkts verantwortlich gemacht werden kann.
- EU Notified Body und UK Approved Body: Mit Ausnahme von Medizinprodukten mit einem geringen Risiko ist für den Zertifizierungsprozess die Einschaltung einer unabhängigen autorisierten Stelle erforderlich. Für die Europäische Union gibt es verschiedene Behörden in den Mitgliedsstaaten. Für Großbritannien müssen die Zertifizierungsstellen, die UK Approved Bodies, ihre Benennung bei der Medicines and Healthcare products Regulatory Agency (MHRA) beantragen.
- MHRA-Registrierung: Alle Medizinprodukte, einschließlich In-vitro-Diagnostika (IVDs), maßgefertigte Produkte und Systeme oder Behandlungseinheiten, müssen bei der MHRA registriert werden.
Es ist zu beachten, dass sich die gesetzlichen Anforderungen ändern können, und es ist ratsam, die jeweils aktuellen Vorschriften der Europäischen Union und des Vereinigten Königreichs zu konsultieren, um die genauen Unterschiede zwischen der EU MDR / EU IVDR und der UK MDR zu kennen.
Übergangsfrist bzw. Übergangsregelungen
Gemäß der ursprünglichen Gesetzgebung sollte die Übergangsfrist zur UKCA-Zertifizierung am 01. Juli 2023 enden. Im Frühjahr 2023 beschloss die britische Regierung jedoch, die Frist für die Zulassung von CE-gekennzeichneten Medizinprodukten auf dem Markt in Großbritannien zu verlängern. Damit soll eine Verknappung dieser wichtigen Waren vermieden werden.
Das bedeutet, dass Hersteller von Medizinprodukten bis zu dem jeweiligen festgelegten Datum sowohl die UKCA- als auch die CE-Kennzeichnung für ihre Produkte verwenden können, um diese in Großbritannien auf den Markt zu bringen.
Für die einzelnen Produkttypen gelten unterschiedliche Fristen:
UKCA-Kennzeichnung
Da die UKCA-Kennzeichnung von der CE-Kennzeichnung zu unterscheiden ist, gelten für die Kennzeichnung separate Regeln. Hersteller müssen die UKCA-Kennzeichnung gut sichtbar, leserlich und haltbar am Produkt anbringen. Alternativ kann der Gesetzgeber zulassen, dass die UKCA-Kennzeichnung auf einem Etikett am Produkt, auf der Produktverpackung oder auf einem Begleitdokument angebracht wird. Sofern erforderlich, muss auch die Kennnummer der im Vereinigten Königreich anerkannten Stelle (UK Approved Body) auf dem Etikett angegeben werden.
Name und Anschrift der UK Responsible Person müssen auf dem Produktetikett, der äußeren Verpackung oder der Gebrauchsanweisung aufgeführt werden. Wo diese Angabe zu finden ist, hängt davon ab, an welcher Stelle die UKCA-Kennzeichnung angebracht wurde. Diese Regel gilt auch für Produkte mit doppelter Kennzeichnung.
Doppelte Kennzeichnung wird bis zum Ende der Übergangszeit weiterhin akzeptiert. In diesen Fällen muss die Kennnummer der Benannten Stelle (EU) oder der im Vereinigten Königreich anerkannten Stelle (UK Approved Body) auf dem Produktetikett angegeben werden.
Weitere Informationen: https://www.gov.uk/guidance/using-the-ukca-marking
Klassifizierung von Medizinprodukten

Vor Beginn des Konformitätsbewertungsverfahrens muss jeder Hersteller den Verwendungszweck seiner Medizinprodukte festlegen und sie auf der Grundlage der UK MDR, Anhang IX der Richtlinie 93/42 klassifizieren. Das Klassifizierungssystem basiert auf dem Grad des Risikos, das ein Produkt für einen Patienten darstellt, sowie auf seinem Verwendungszweck.
Es gibt vier Klassen:
- Klasse I – Medizinprodukte, die ein geringes bis mittleres Risiko für den Patienten darstellen
- Klasse IIa – Medizinprodukte, die ein mittleres Risiko für den Patienten darstellen
- Klasse IIb – Medizinprodukte, die ein mittleres bis hohes Risiko für den Patienten darstellen
- Klasse III – Medizinprodukte, die ein hohes Risiko für den Patienten darstellen
Das Klassifizierungssystem basiert auf 18 Regeln, die ebenfalls in dem genannten Anhang beschrieben sind. Die Klassifizierungsregeln sind in vier Kategorien eingeteilt:
- Nicht invasive Produkte, die nicht in den menschlichen Körper eindringen
- Invasive Produkte, die in den menschlichen Körper eindringen (einschließlich chirurgisch-invasiver Produkte)
- Zusätzliche Regeln gelten für aktive Produkte, die eine externe Energiequelle benötigen
- Besondere Vorschriften für alle sonstigen Produkte, die nicht in die anderen Kategorien fallen
Für Medizinprodukte der Klasse I
Produkte wie Wundpflaster oder In-vitro-Diagnostika stellen in der Regel ein geringes Risiko für die Sicherheit eines Patienten dar. Daher werden solche Produkte als Klasse I (Medizinprodukte mit geringem Risiko) eingestuft. Hersteller können die Konformität ihres Produkts selbst erklären, bevor sie ein UKCA-Zeichen anbringen, und sie können ihr Produkt auf den Markt bringen, ohne eine im Vereinigten Königreich anerkannte Stelle (UK Approved Body) einzuschalten.
Medizinprodukte der Klasse I, die steril sind oder eine Messfunktion haben, sind eine Ausnahme von dieser Regel. Für diese Produkte ist die Einschaltung einer vom Vereinigten Königreich anerkannten Stelle (UK Approved Body) zur Durchführung einer externen Konformitätsbewertung erforderlich.
Technische Dokumentation
Gemäß der UK MDR gelten besondere Anforderungen an die technische Dokumentation von Medizinprodukten. Diese Anforderungen sind in den offiziellen Leitlinien der britischen Regierung zur UKCA-Kennzeichnung, -Konformitätsbewertung und -Dokumentation detailliert dargelegt. Die Dokumentation muss die Konformität mit den in den Vorschriften verankerten Anforderungen belegen. Sie sollte Informationen über die Auslegung und den Herstellungsprozess des Produkts, die Leistungsdaten und die Risikobewertung enthalten. Darüber hinaus sollte die technische Dokumentation Informationen über Qualitätsmanagementsysteme, Kennzeichnung und die Gebrauchsanweisung enthalten.
Konformitätsbewertungsverfahren für Medizinprodukte und In-vitro-Diagnostika im Vereinigten Königreich
In manchen Fällen (z. B. bei Produkten mit einem höheren Risiko) ist die Konformitätsbewertung obligatorisch für das Inverkehrbringen eines Medizinprodukts im Vereinigten Königreich. Die Hersteller von Medizinprodukten investieren viel Zeit und Geld in die Entwicklung ihrer Produkte. Wenn diese Produkte nicht den gültigen Anforderungen entsprechen, können teure und zeitaufwändige Änderungen erforderlich werden. Dies verursacht nicht nur zusätzliche Kosten, sondern verzögert auch die Markteinführung.
Die UK MDR legt fest, welches Verfahren für das jeweilige Medizinprodukt gilt. Bei Produkten mit geringerem Risiko, wie z. B. bei Produkten der Klasse I, können Hersteller die Konformitätsbewertung in der Regel selbst vornehmen. Dazu gehören die Durchführung einer Risikoanalyse, die Implementierung eines Qualitätsmanagementsystems und die Zusammenstellung eines technischen Dossiers, das die Übereinstimmung des Produkts mit den einschlägigen Anforderungen nachweist.
Für Produkte mit höherem Risiko, wie Produkte der Klassen IIa, IIb und III, ist das Konformitätsbewertungsverfahren komplexer. Es umfasst in der Regel eine Bewertung durch eine im Vereinigten Königreich anerkannte Stelle (UK Approved Body), wie z. B. TÜV Rheinland UK Ltd.
Die Aufgabe dieser Stelle ist es, unabhängig zu überprüfen, ob der Hersteller die Anforderungen der UK MDR erfüllt. Dazu gehört die Prüfung der technischen Unterlagen des Herstellers, die Auditierung seiner Fertigungsanlagen und gegebenenfalls die Durchführung von Prüfungen an dem Produkt.
Der Geltungsbereich der Benennung von TÜV Rheinland UK Ltd. ist auf bestimmte Konformitätsbewertungsverfahren beschränkt. Das bedeutet, dass TÜV Rheinland UK Ltd. entsprechend seiner Benennung befugt ist, Konformitätsbewertungen für bestimmte Arten von Medizinprodukten durchzuführen. Der Geltungsbereich ist breit, und TÜV Rheinland UK Ltd. kann eine Vielzahl von Medizinprodukten und In-vitro-Diagnostika zertifizieren.
Nach dem erfolgreichen Abschluss des Konformitätsbewertungsverfahrens kann der Hersteller die UKCA-Kennzeichnung auf seinem Produkt anbringen. Diese Kennzeichnung signalisiert, dass das Produkt den geltenden Vorschriften des Vereinigten Königreichs entspricht und dort in Verkehr gebracht werden kann.
MHRA-Registrierung
Alle Medizinprodukte, einschließlich IVDs, maßgefertigte Produkte und Systeme oder Behandlungseinheiten, die in Großbritannien (England, Schottland, Wales) auf den Markt gebracht werden, müssen seit 2021 bei der Medicines and Healthcare products Regulatory Agency (MHRA) registriert werden.
Der Nachweis der Konformität ist Voraussetzung für die Registrierung. Für Hersteller und Vertriebshändler, die nicht im Vereinigten Königreich ansässig sind, ist die Registrierung Aufgabe der UK Responsible Person.
Dies gilt für folgende Produkte:
- Produkte der Klassen I, IIa, IIb oder III, die Sie hergestellt haben
- Produkte der Klassen I, IIa, IIb oder III, die Sie überarbeitet oder mit Ihrem eigenen Namen neu gekennzeichnet haben
- Jedes System oder jede Behandlungseinheit, die mindestens ein Medizinprodukt enthält
- Maßgefertigte Produkte
- IVDs, die Sie hergestellt haben
- IVDs, die eine Leistungsbewertung durchlaufen
Für neue Registrierungen oder Änderungen bestehender Registrierungen erhebt die Organisation eine Standardgebühr von £100.
In Nordirland
Unter bestimmten Bedingungen muss die MHRA über die Konformität mit der UK MDR informiert werden, wenn Sie Ihr Produkt auf den Markt in Nordirland bringen. Die konkreten Anforderungen hängen von bestimmten Faktoren ab, wie dem Standort des Herstellers, dem Standort des bevollmächtigten Vertreters und der Klassifizierung des Produkts.
Bei maßgefertigten Medizinprodukten sind Sie verpflichtet, die MHRA innerhalb von 28 Tagen nach der Markteinführung in Nordirland zu informieren.
Wenn Sie ein in Nordirland ansässiger Hersteller sind und Ihr Produkt bereits bei der MHRA für den Markt in Nordirland registriert haben, können Sie es anschließend in Großbritannien auf den Markt bringen, ohne es zusätzlich in Großbritannien registrieren zu müssen.
Dies gilt jedoch in den folgenden Fällen nicht für Hersteller, die Medizinprodukte der Klasse I oder allgemeine IVDs (mit Ausnahme von Produkten zur Eigenanwendung) in Nordirland auf den Markt bringen:
- Der Hersteller ist in der EU oder im EWR ansässig.
- Der Hersteller ist außerhalb Nordirlands, der EU oder des EWR ansässig, hat jedoch einen Bevollmächtigten mit Sitz in der EU benannt.
Mehr über die Zulassung von Medizinprodukten für den UK-Markt.
Die Rolle von TÜV Rheinland als UK Approved Body
Die TÜV Rheinland Group ist ein weltweit führender Anbieter von unabhängigen Prüfdienstleistungen und ein bewährter Partner für die Sicherung der Qualität und Sicherheit von Produkten. Im Zusammenhang mit den Vorschriften für Medizinprodukte im Vereinigten Königreich spielt TÜV Rheinland UK Ltd. als eine im Vereinigten Königreich anerkannte Stelle (UK Approved Body) eine zentrale Rolle bei den für die UKCA-Zertifizierung erforderlichen Konformitätsbewertungsverfahren.
Als UK Approved Body sind wir von der britischen Regierung autorisiert, Konformitätsbewertungsverfahren durchzuführen, um zu verifizieren, dass die Hersteller die Anforderungen der UK MDR erfüllen. Dies umfasst eine gründliche Überprüfung der technischen Dokumentation des Herstellers, die Auditierung seiner Fertigungsanlagen und gegebenenfalls die Durchführung von Prüfungen am Produkt.
Durch eine Partnerschaft mit uns profitieren Hersteller von unserer umfassenden Erfahrung und Expertise auf dem Gebiet der Regelungen für Medizinprodukte.
Selbstverständlich bieten wir auch Prüfungen von Medizinprodukten sowie Zertifizierungen gemäß den EU-Verordnungen an. Wir sind außerdem eine EU-Benannte Stelle und führen Dienstleistungen gemäß (EN) ISO 13485, EU MDR und EU IVDR durch und bieten zudem die MDSAP-Zertifizierung als Auditierungsorganisation an. Unser Market Access Service ist der letzte Grundpfeiler unserer Komplettlösung aus einer Hand, die Ihnen den Zugang zu internationalen Märkten für Ihre Medizinprodukte ermöglicht.
Die Umstellung von CE- auf UKCA-Kennzeichnung
Die Umstellung von der CE- auf die UKCA-Kennzeichnung erfordert das Verständnis der Anforderungen der UK MDR, die Durchführung einer Gap-Analyse, um Unterschiede zwischen den Anforderungen an die CE- und die UKCA-Kennzeichnung zu ermitteln, die Umsetzung aller erforderlichen Änderungen, die Durchführung des erforderlichen Konformitätsbewertungsverfahrens und die Anbringung der UKCA-Kennzeichnung am Produkt.
Zunächst müssen Hersteller die Anforderungen für die UKCA-Kennzeichnung, so wie sie in der UK MDR dargelegt sind, vollkommen verstehen. Dazu gehören die Anforderungen an die Auslegung und Herstellung des Produkts sowie das spezifische Konformitätsbewertungsverfahren, das angewandt werden muss.
Anschließend sollten die Hersteller eine Gap-Analyse durchführen, um eventuelle Unterschiede zwischen den Anforderungen der CE- und der UKCA-Kennzeichnung zu ermitteln. Auf diese Weise kann festgestellt werden, welche Änderungen vorgenommen werden müssen, um die Anforderungen der UKCA-Kennzeichnung zu erfüllen.
Sobald die notwendigen Änderungen ermittelt wurden, muss der Hersteller diese implementieren und das erforderliche Konformitätsbewertungsverfahren durchführen. Dazu kann eine Bewertung durch eine im Vereinigten Königreich anerkannte Stelle (UK Approved Body) wie TÜV Rheinland UK Ltd. notwendig sein.
Nach dem erfolgreichen Abschluss der Konformitätsbewertung und nach der Ausstellung des UKCA-Zertifikats können Hersteller ihr Produkt bei der MHRA registrieren lassen und die UKCA-Kennzeichnung am Produkt anbringen.
Bevorstehende Änderungen
Für die Zukunft hat die britische Regierung Pläne zur Implementierung von Vorschriften angekündigt, die den derzeitigen Rechtsrahmen für Medizinprodukte im Vereinigten Königreich erheblich reformieren werden. Der Plan für diese Reform wurde in der Antwort der Regierung auf die Konsultation über die zukünftige Regulierung von Medizinprodukten im Vereinigten Königreich im Jahr 2021 dargelegt. Ziel ist es, eine schrittweise und angemessene Umsetzung des künftigen Rechtsrahmens zu gewährleisten, wobei die Systembereitschaft im Vordergrund stehen und das Risiko von Versorgungsengpässen für Patienten minimiert werden soll. Dieser Leitfaden wurde aktualisiert, und es wurde festgelegt, dass die Hauptaspekte der künftigen Rechtsvorschriften für Medizinprodukte nun ab dem 1. Juli 2025 gelten sollen.
Darüber hinaus beabsichtigt die Regierung, noch im Laufe von 2023 Rechtsvorschriften einzuführen, um im Vorfeld der zukünftigen umfassenden Regulierungsvorschriften strengere Anforderungen an die Überwachung nach dem Inverkehrbringen durchzusetzen. Dies zeigt das Bestreben der Regierung, die Sicherheit von Patienten als einen integralen Bestandteil der künftigen Vorschriften für Medizinprodukte verbessern zu wollen. Diese Anforderungen an die Überwachung nach dem Inverkehrbringen sollen ab Mitte 2024 in Kraft treten.
Erfahren Sie mehr.



{kind=link}
{kind=link}
Sichern Sie sich jetzt den Zugang zu internationalen Märkten
