

Your Comprehensive Guide to UK Medical Devices Regulations
Medical devices are very diverse. They cover a wide range of products, from surgical masks over ventilators to cardiac catheters. As different as these products are, they have one thing in common. For the protection of the patient, they are subject to the highest quality standards and specific legal requirements depending on their classification.
Understanding the regulatory framework of a specific sales market is crucial for manufacturers who seek to launch their product. On this website, we focus on the regulatory landscape for medical devices in the United Kingdom. Since leaving the European Union, the UK has undergone significant changes, particularly with the introduction of the UKCA (United Kingdom Conformity Assessed) marking and the UKNI (United Kingdom Northern Ireland) marking.
UKCA – four letters that open a new door
The UK government introduced the UKCA mark as clear proof that a product complies with the applicable legislation. It covers most products that previously required the CE marking. That´s why for many product groups, the legal requirements of the UKCA mark still reflect the CE certification requirements. However, more and more specific regulations are coming into effect in the UK that differ from the EU regulations and need to be taken into account by manufacturers and retailers.
For manufacturers of medical devices, in-vitro diagnostic medical devices and active implantable medical devices, this marking is of paramount importance as it indicates that their product complies with the applicable UK regulatory requirements. For this product group, a UKCA marking is mandatory and sometimes even requires involving a UK Approved Body.
The requirements are defined in The Medical Device Regulations 2002 (SI 2002 No 618, as amended) (UK MDR). The underlying European directives referenced in the UK MDR cover various aspects of the device requirements, including its design, production, and labelling, as well as the information to be supplied with the device. We will take a closer look at the UK MDR in the section below.
Important to consider is that the UK government extended the transition period (transitional arrangements) from CE to UKCA marking for many product groups to avoid shortages of medical devices on the British market. In the section “Transitional Arrangements” we provide details for different types of medical devices. Nevertheless, all medical devices placed on the Great Britain market (England, Scotland, Wales) must be registered with the Medicines and Healthcare products Regulatory Agency (MHRA) as of 2021. Also, certain medical devices which are placed in the Northern Ireland (including IVDs, custom-made devices and systems and procedure packs) must be registered with the MHRA.
The UKCA certification scheme – Who is involved?
MHRA
The Medicines and Healthcare products Regulatory Agency is the government agency responsible for ensuring that medical devices and other medical products function and are acceptably safe. The MHRA plays a vital role in the regulation of medical devices in the UK, overseeing the entire lifecycle from product design, manufacturing, clinical evaluation to post-market surveillance.
The agency is also responsible for designating and monitoring organizations that provide certification according to UK MDR. These are the so-called UK Approved Bodies.
Note: Accreditations for other product groups like PPE, machinery, toys, etc. will be issued by UKAS, the United Kingdom Accreditation Service.
UK Approved Body
A UK Approved Body is an organization that is designated by the MHRA to carry out a specific conformity assessment, in order to ensure compliance with regulatory standards. The designation of an organization as an UK Approved Body typically involves a formal application and assessment process, where the organization must demonstrate its competence, expertise, resources, and ability to fulfil the designated functions or responsibilities.
We are happy to announce that TUV Rheinland UK Ltd. Is designated by the MHRA as an UK Approved Body under UK MDR.
Note: Former Notified Bodies based in the UK have become UK Approved Bodies. They are no longer able to issue CE certificates for the EU market because the EU does not recognize them.
UK Responsible Person
The UK MDR introduces the role of a UK Responsible Person who acts on behalf of the manufacturer placed outside of the UK.
The purpose is to ensure that there is a legal entity within the UK that can be held accountable for the compliance of the device. Therefore, manufacturers based outside the UK need to appoint a UK Responsible Person by a written mandate, which must outline the specific tasks for which they are responsible. This is required in order to place their product on the UK market.
The key responsibilities are:
- To ensure that the conformity assessment procedure has been carried out
- To ensure that the technical documentation and declaration of conformity are available upon request by the MHRA
- To respond to all requests by the MHRA
- To keep a register of complaints, non-conforming devices, and product recalls and to provide information about it
- To report serious incidents to the MHRA
However, the manufacturer remains ultimately responsible for the compliance of their device, even when tasks are delegated to a UK Responsible Person.
Manufacturer
The company (manufacturer or importer) of a medical device is obligated to ensure that the product has been designed and manufactured in accordance with the relevant requirements of the relevant legislation. That means he is also responsible for carrying out a conformity assessment, regardless of whether a self-declaration is sufficient, or a mandatory third-party conformity assessment is required. The manufacturer must draw up a technical documentation and a UK declaration of conformity and keep both records for 10 years. He is also required to take care of suitable labelling.
Note: This also applies to importers or distributors who distribute a product under their name and trademark as they are usually considered the manufacturer.
Find out more about the responsibilities of different parties on the following UK government website.

Where does the UKCA mark apply?
At the first glance, it seems obvious, that the UKCA mark applies for goods sold in the United Kingdom. We would like to point out some facts that often lead to confusion.
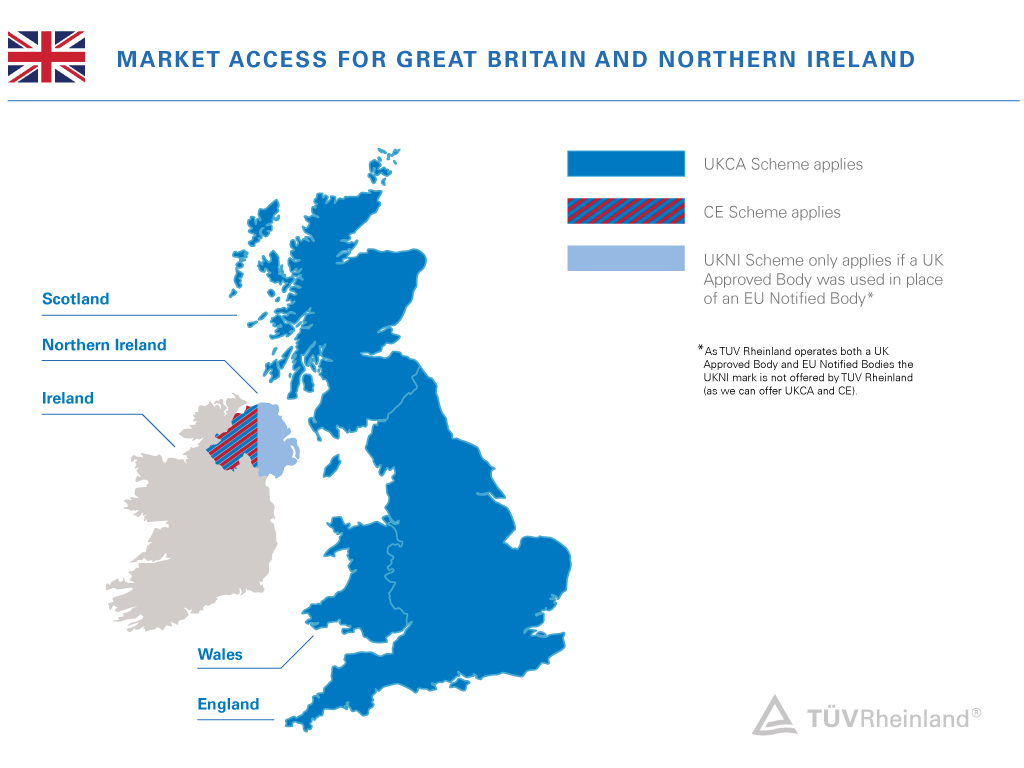
When speaking about the United Kingdom, it means the union of four countries: England, Wales, Scotland, and Northern Ireland. Great Britain, on the other hand, only covers England, Wales, and Scotland.
For Northern Ireland with its domestic border to Ireland (and thus the European Union), special rules apply that are defined in the Northern Ireland Protocol.
As of 2022, both regulations, the EU MDR (Regulation 2017/745) and EU IVDR (Regulation 2017/746) apply in Northern Ireland.
That means, the UK MDR and the UKCA certification scheme only apply to countries in Great Britain, and the sales market in Northern Ireland requires a CE marking!
CE and UKNI marking for Northern Ireland
Marketing a product in Northern Ireland requires the CE mark, which can be affixed after self-declaration or issued by an EU Notified Body by an EU member state authority according to the EU MDR or EU IVDR. If a device manufacturer uses an EU-recognized Notified Body for mandatory third-party conformity assessment, the CE marking on its own is sufficient to place a device on the Northern Ireland market.
But, if a UK Approved Body undertakes the mandatory third-party conformity assessment, the UKNI indication is required. In these cases, device manufacturers must not apply the UKNI indication on its own - it must always accompany a CE marking. Goods bearing the combined “CE & UKNI” marking will not be accepted on the EU market.
Please note that the rules and exceptions for Northern Ireland have changed very dynamically in the course of the negotiations between the United Kingdom and the European Union.
Here are some current differences between Great Britain and Northern Ireland:
- Dual marking: In Northern Ireland, both the UKNI marking and the CE marking are accepted. While a CE mark alone is sufficient to market a product, the UKNI mark always must be accompanied by the CE mark.
- UKNI marking: Some products that are subject to specific EU legislation and designated as "relevant Northern Ireland goods" require the UKNI marking. This marking is used for products that are manufactured in or imported into Northern Ireland and are intended for sale in Northern Ireland or both Northern Ireland and the EU market.
- Conformity Assessment Bodies: Manufacturers in Northern Ireland can continue to use EU-recognized conformity assessment bodies (CABs). This is important for manufacturers that require third-party conformity assessment for medical devices of higher risks.
- Northern Ireland Declaration of Conformity: Manufacturers placing products on the Northern Ireland market should issue a Northern Ireland Declaration of Conformity. This declaration should state that the product complies with the applicable UK or EU regulations, depending on the intended market.
- MHRA registration: All medical devices distributed in the UK (Great Britain or Northern Ireland) require an MHRA registration. All custom-made devices must be registered with the MHRA within 28 days of being made available on the Northern Ireland market.
Understanding UK MDR 2002
As mentioned above, the Medical Devices Regulations 2002 (SI 2002 No 618, as amended) (UK MDR ) is a key piece of legislation that governs the production and distribution of medical devices in the UK. It is crucial for manufacturers to understand this regulation as it sets out the requirements that must be met to legally place a medical device on the market in the UK.
The UK MDR gave effect in UK law to three directives:
- Medical Devices Directive 93/42/EEC (MDD)
- Active Implantable Medical Devices Directive 90/385/EEC (AIMDD)
- In-Vitro Diagnostic Medical Devices Directive 98/79/EC (IVDD)
Therefore, when speaking of medical devices in the context of UKCA certification, this also includes in-vitro diagnostic medical devices and active implantable medical devices.
Since the UK MDR is based on these European directives, the UKCA marking requirements are based on the requirements derived from the EU legislation. However, with the UK’s withdrawal from the European Union, the UK MDR has been amended to apply independently.
The UK MDR covers a wide range of topics, including the classification of medical devices, the conformity assessment procedures, the role of UK Approved Bodies, post-market surveillance, and the requirements for clinical evaluation. It also introduces several key terms and abbreviations that are used throughout the regulation.
One of the key aspects of the UK MDR are the conformity assessment procedures. Manufacturers must follow these processes to demonstrate that their medical device complies with the requirements of the regulation. Depending on the medical device and its classification, a specific procedure needs to be selected. Medical devices classified as higher risk generally requiring a more rigorous assessment.
The UK MDR also outlines the requirements for the performance of audits, the qualification of personnel, and the control of documents and records. These requirements are designed to ensure that manufacturers have robust quality management systems in place, and that they are maintaining a high level of control over their processes and products.
Most important differences between EU MDR and UK MDR
Although the UK MDR is based on common directives, there are some important differences between these two regulations. Manufacturers who supply both markets must follow the specifications of both regulations very carefully and apply for separate certifications under the respective regulations.
Here are the biggest differences:
- Scope: Basically, the EU MDR and EU IVDR apply in all European Union member states and Northern Ireland, while the UK MDR applies only in the Great Britain. Please note that the transition period is still ongoing. CE marking will continue to be recognized and accepted in the UK. The expiry dates depend on the medical device classification.
- Certification: Different conformity assessment procedures apply to the certification of medical devices. The EU MDR/EU IVDR uses the EU Notified Body, while the UK MDR uses the UK Approved Body for relevant conformity assessment procedures.
- Product groups: The UK MDR includes general medical devices, active implantable medical devices and in-vitro diagnostic medical devices (IVDs). For the EU market, the requirements are defined separately in the EU MDR and EU IVDR.
- UKCA Marking: British market requires a different marking on the product and/or packaging. The number of the UK Approved Body must appear on the device or product label. To ease the transition for manufacturers, devices with a CE mark do not have to be re-labelled, and dual marking will also continue to be accepted until the end of the transition period (transitional arrangements).
- UKCA Responsible Person: We already explained above that non-UK manufacturers need to appoint a UK responsible person in order to ensure that there is a legal entity within the UK that can be held accountable for the compliance of the device.
- EU Notified Body and UK Approved Body: Except for low-risk medical devices, the certification process requires involving an independent and authorized third party. For the European Union, there are various authorities in the member states. For Great Britain, certification bodies, the UK Approved Bodies, need to apply for designation at the Medicines and Healthcare products Regulatory Agency (MHRA).
- MHRA registration: All medical devices, including in-vitro diagnostic medical devices (IVDs), custom-made devices and systems or procedure packs, need to be registered with the MHRA.
It is important to note that regulatory requirements are subject to change, and it is advisable to consult the respective current European Union regulations and UK regulations for the exact differences between the EU MDR / EU IVDR and the UK MDR.
Transition period called Transitional Arrangements
According to the original legislation, the transition period to the UKCA certification was to end on July 01, 2023. In the spring of 2023, the UK government decided to extend the period of acceptance of CE marked medical devices on the Great Britain market. This is to avoid a shortage of the important goods.
This means that until the specified date, manufacturers of medical devices can use both, the UKCA and the CE mark, to place their products on the Great Britain market.
Different timelines apply to the individual product types:
UKCA Marking
Since the UKCA mark is to be distinguished from the CE mark, it follows its own rules for labelling. Manufacturers need to place the UKCA marking visibly, legibly, and indelibly on the product. Legislation may allow the UKCA marking to be alternatively affixed to a label placed on the product, to the product’s packaging or to an accompanying document. Where relevant, the number of the UK Approved Body must also appear on the label.
Regarding the UK responsible person, the name and address needs to be included on the product labelling, the outer packaging, or the instructions for use. It depends on the spot where the UKCA marking has been affixed. This rule applies also to dual-marked products.
Dual marking will continue to be accepted until the transition period ends. In these cases, the number of the Notified Body (EU) or Approved Body (UK) must appear on the device or product label.
More information: https://www.gov.uk/guidance/using-the-ukca-marking
Classification of medical devices

Before starting the conformity assessment procedure, every manufacturer must define the intended purpose of their medical devices and classify them based on UK MDR, Annex IX of Directive 93/42. The classification system is based on the level of risk a product poses to the patient and on the intended use.
There are four classes:
- Class I – Medical devices that pose a low to moderate risk to the patient
- Class IIa – Medical devices that pose a moderate risk to the patient
- Class IIb – Medical devices that pose a moderate to high risk to the patient
- Class III – Medical devices that pose a high risk to the patient
The classification system is based on 18 rules, also described in the mentioned Annex. The applicable rules are organized in four categories:
- Non-invasive devices that do not penetrate the human body
- Invasive devices that penetrate the human body (incl. surgically invasive devices)
- Additional rules apply to active devices that require an external energy source
- Special rules for all other devices falling outside of the other categories
For Class I medical devices
Products such as adhesive bandages or in-vitro diagnostics are usually of low risk for a patient’s safety. Therefore, such products are classified as Class I (low risk medical devices). Manufacturers can self-declare conformity before affixing a UKCA mark and place their device on the market without involving an UK Approved Body.
Class I medical devices that are sterile or have a measuring function are an exception from this rule. They require the involvement of a UK Approved Body to undertake a third-party conformity assessment.
Technical Documentation
According to the UK MDR, specific conditions apply to the technical documentation of medical devices. These requirements are outlined in detail in the official guidance provided by the UK government on UKCA marking, conformity assessment, and documentation. The documentation must demonstrate conformity with the requirements specified in the regulations. It should include information such as the device's design and manufacturing process, performance data, and risk assessment. Additionally, the technical documentation should cover aspects related to quality management systems, labelling, and instructions for use.
Conformity Assessment Procedures for Medical Devices and In-Vitro Diagnostic Medical Devices in the UK
In some cases (i.e. higher risk devices) the conformity assessment is a mandatory step in the process of placing a medical device on the market in the UK. Medical device manufacturers invest a lot of time and money in the development of their products. If they do conform with relevant requirements, expensive and time-consuming changes may be necessary. This not only results in additional costs, but also delays the market launch.
The UK MDR specifies which process applies to the medical device in question. For lower-risk devices, such as Class I devices, manufacturers can typically carry out the conformity assessment themselves. This involves conducting a risk analysis, implementing a quality management system, and compiling a technical file that demonstrates the device’s compliance with the relevant requirements.
For higher-risk devices, such as Class IIa, IIb, and III devices, the conformity assessment procedure is more complex. It typically involves an assessment by a UK Approved Body, such as TÜV Rheinland UK Ltd.
The role of the UK Approved Body is to verify independently that the manufacturer complies with the requirements of the UK MDR. This involves reviewing the manufacturer’s technical documentation, auditing their manufacturing facilities, and potentially (if required) conducting tests on the device.
The scope of designation for TÜV Rheinland UK Ltd. is limited to certain conformity assessment procedures. This means that TÜV Rheinland UK Ltd. is authorized to conduct conformity assessments for certain types of medical devices, as specified in its designation. This is large scope and TUV Rheinland UK Ltd. can certify various medical devices and In-vitro diagnostic medical devices.
Once the conformity assessment procedure has been successfully completed, the manufacturer can affix the UKCA marking to their device. This marking signifies that the device complies with the applicable UK regulatory requirements and can be placed on the UK market.
MHRA Registration
All medical devices including IVDs, custom-made devices and systems or procedure packs placed on the Great Britain market (England, Scotland, Wales) must be registered with the Medicines and Healthcare products Regulatory Agency (MHRA) as of 2021.
Proof of conformity is a prerequisite for registration. For manufacturers and distributors not based in the UK, registration is a task for the UK responsible person.
This covers the following products:
- Class I, IIa, IIb or III devices you have manufactured
- Class I, IIa, IIb or III devices you have refurbished or re-labelled with your own name
- Any system or procedure pack containing at least one medical device
- Custom-made devices
- IVDs you have manufactured
- IVDs undergoing performance evaluation
For new registrations or changes to existing registrations, the organization charges a £100 standard fee.
In Northern Ireland
Under certain conditions, compliance with the UK MDR necessitates notifying the MHRA when introducing your device to the Northern Ireland market. The specific requirements vary depending on factors such as the manufacturer's location, the location of the Authorized Representative, and the device's classification.
For custom-made devices, it is mandatory to inform the MHRA within 28 days of making them available on the Northern Ireland market.
If you are a manufacturer based in Northern Ireland and have already registered your device with the MHRA for the Northern Ireland market, you can subsequently place it on the Great Britain market without undergoing any additional registration in Great Britain.
However, this requirement does not apply to manufacturers who place Class I medical devices or general IVDs (excluding self-testing ones) on the Northern Ireland market in the following cases:
- The manufacturer is located in the EU or EEA.
- The manufacturer is located outside Northern Ireland, the EU, or EEA but has designated an Authorized Representative based in the EU.
More about the registration of medical devices for the UK market.
The role of TÜV Rheinland as a UK Approved Body
TÜV Rheinland Group is a global leader in independent inspection services and a trusted partner for ensuring the quality and safety of products. In the context of UK medical devices regulations, TUV Rheinland UK Ltd. serves as a UK Approved Body, playing a crucial role in the conformity assessment procedures required for the UKCA certification.
As a UK Approved Body, we are authorized by the UK government to conduct conformity assessment procedures to verify manufacturers compliance with the requirements of the UK MDR. This involves a thorough review of the manufacturer’s technical documentation, audits of their manufacturing facilities, and potentially (if required) conducting tests on the device.
By working with us, manufacturers can benefit from our extensive experience and expertise in the field of medical device regulations.
Of course, we also offer medical device testing as well as certification for the European Union Regulations. We are also an EU Notified Body and conduct services according to (EN) ISO 13485, EU MDR, EU IVDR and as well we offer MDSAP certification as the auditing organization. Our Market Access Service is the last pillar to offer you a comprehensive one-stop solution for accessing international markets with your medical devices.
How to Transition from CE to UKCA Marking
Transitioning from CE to UKCA marking involves understanding the requirements for UK MDR requirements, conducting a gap analysis to identify any differences between the CE and UKCA marking requirements, implementing any necessary changes, carrying out the required conformity assessment procedure, and affixing the UKCA marking to the device.
First, manufacturers must ensure that they fully understand the requirements for UKCA marking, as outlined in the UK MDR. This includes the requirements for the design and manufacture of the device, as well as the specific conformity assessment procedure that must be followed.
Next, manufacturers should conduct a gap analysis to identify any differences between the CE and UKCA marking requirements. This will help to identify any changes that need to be made in order to comply with the UKCA marking requirements.
Once any necessary changes have been identified, the manufacturer must implement them and carry out the required conformity assessment procedure. This may involve an assessment by a UK Approved Body, such as TÜV Rheinland UK Ltd.
After the conformity assessment has been successfully completed and UKCA certificate issued, manufacturers can register their product with the MHRA and affix the UKCA marking to the device.
Changes around the corner
For the future, the UK government announced plans to implement regulations that will significantly reform the current regulatory framework for medical devices in the UK. The approach to this reform was outlined in their response to the 2021 consultation on the future regulation of medical devices in the UK. The aim is to ensure a phased and proportionate implementation of the future regulatory framework, prioritizing system readiness and minimizing the risk of supply disruption for patients in the UK. This guidance has been updated to indicate that core aspects of the future medical device regime are now scheduled to apply beginning July 1, 2025.
Additionally, the government intends to introduce legislation later in 2023 to enforce enhanced post-market surveillance requirements in advance of the broader future regulatory regime. This demonstrates the government's commitment to improving patient safety as an integral part of the future medical device regulations. These post-market surveillance requirements are expected to come into effect starting mid-2024.
Find out more



{kind=link}
{kind=link}
Achieve international market access now
