Regulamentul (UE) 2017/745 privind dispozitivele medicale (MDR)

|
Regulamentul (UE) 2020/561 al Parlamentului European și al Consiliului din 23 aprilie 2020 de modificare a Regulamentului (UE) 2017/745 privind dispozitivele medicale a fost publicat în data de 24 aprilie 2020 în Jurnalul Oficial al Uniunii Europene! Obiectivul principal este amânarea datei de aplicare din 26 mai 2020 până la 26 mai 2021. O dată cu această amânare au fost adoptate și alte date de aplicare a altor dispoziții. Vă rugăm găsiți pagina oficială a Regulamentului de modificare aici. |
|---|

Un început în conformitate cu noul Regulament UE privind dispozitivele medicale

Producătorii de dispozitive medicale trebuie să se adapteze la noile cerințe impuse de MDR 2017/745, care a intrat în vigoare la 25 mai 2017. A fost publicat și Regulamentul (UE) 2020/561 al Parlamentului European și al Consiliului din 23 aprilie 2020 de modificare a Regulamentului (UE) 2017/745 privind dispozitivele medicale prin care se amână data aplicării pâna la 26 MAI 2021. TÜV Rheinland a apreciat și susține amânarea.
Măsura oferă producătorilor de dispozitive medicale posibilitatea de a concentra pe deplin toate resursele posibile pe lupta împotriva pandemiei COVID-19. Producătorii de dispozitive medicale joacă un rol important, însă se confruntă cu multe provocări. Este extrem de important ca dispozitivele medicale să rămână conforme și să rămână disponibile în UE pentru a evita deficiențele sau întârzierile anumitor dispozitive medicale în aceste circumstanțe unice.
Dincolo de această situație, TÜV Rheinland ar dori să-și încurajeze clienții să țină legătura cu noi pentru a continua planificarea activităților de punere în aplicare a Regulamentului privind dispozitivele medicale, tinând cont bineînțeles și de noua dată de aplicare, 26 mai 2021.
Auditurile efectuate în conformitate cu MDR asigură accesul pe piețele UE
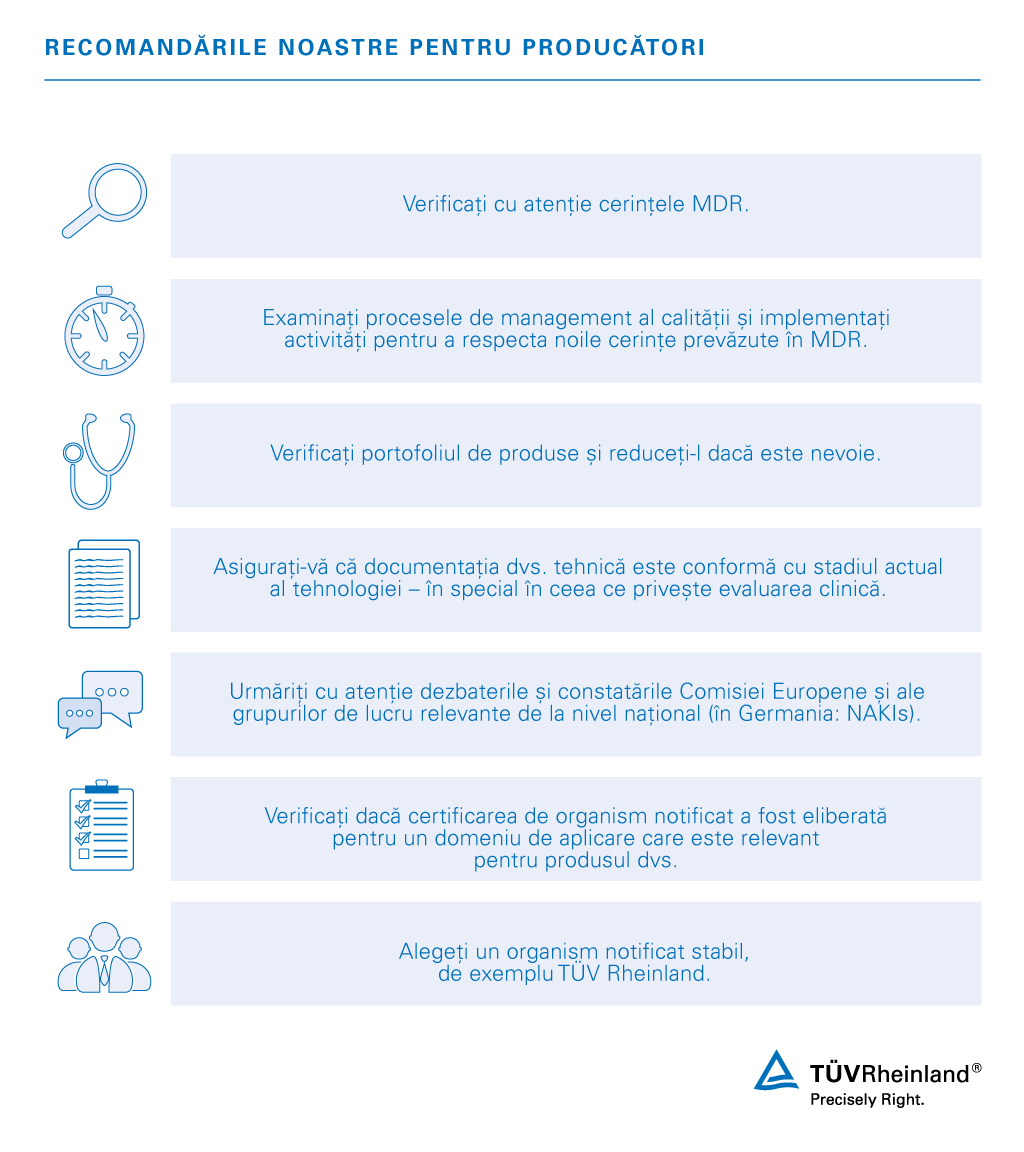
Odată cu înlocuirea prin MDR 2017/745 a normelor existente care reglementează dispozitivele medicale din UE, producătorii vor fi în curând obligați să își reevalueze produsele în vederea asigurării conformității. O procedură de evaluare a conformității cu MDR oferă companiilor posibilitatea de a obține certificarea necesară pentru a introduce produsele pe piața europeană. Evaluarea conformității pentru acces, condiția de bază pentru succesul reglementării.
În cadrul procesului dvs. de asigurare a conformității vă putem oferi recomandări, informații și, în cele din urmă, putem efectua orice verificare și audit a documentației tehnice necesare. Suntem sprijiniți de o rețea mondială de specialiști cu experiență în acest sector industrial și dispunem de facilități și oferim un punct unic de contact pentru serviciile aferente dispozitivelor medicale.
Evaluarea conformității dispozitivului medical și alte aspecte
În prezent, serviciile noastre se concentrează asupra perioadei de tranziție și asupra calendarului pentru asigurarea conformității cu MDR 2017/745. Experții noștri vă pot oferi asistență pentru a respecta termenele și pentru a aborda toate problemele relevante pentru menținerea accesului dispozitivelor dvs. medicale pe piețele europene.
Partenerul dvs. de încredere pentru gestionarea înlocuirii directivei privind dispozitivele medicale
În calitate de organizație de testare și certificare și de specialist pentru accesul pe piață la nivel mondial, oferim servicii complete în domeniul industriei dispozitivelor medicale, toate în același loc. Pe lângă faptul că vă permite să gestionați tranziția pentru a vă adapta la noul Regulament (UE) privind dispozitivele medicale, serviciile noastre includ auditul sistemului de management al calității pentru producătorii de dispozitive medicale, furnizori și birourile de vânzări, precum și testarea dispozitivelor medicale. Suntem inovatori și pregătiți pentru viitoarele provocări în domeniul digitalizării, precum conexiunile wireless, telemedicina, aplicațiile în domeniul medical securitate cibernetică, protecția datelor cu caracter personal și multe altele.
Consultați un expert pentru a începe în timp util asigurarea conformității cu MDR.
Întrebări și răspunsuri cu privire la noul Regulament (UE) 2017/745 privind dispozitivele medicale (MDR)
Informații suplimentare disponibile pentru descărcare
| FAQ – Întrebări frecvente cu privire la MDR | 1 MB | Descărcare |
Contact

/tuv-rheinland-de19_p05_ivd09-lp_core_4_3.jpg)
{kind=link}